-
PROVIDERS
New MRD Medicare Coverage for Select Indications*
*When coverage criteria are met. Additional criteria and exceptions for coverage may apply.
-
LIFE SCIENCES
ENROLL NOW
Tempus’ patient-derived organoid screens
Evaluate the efficacy of your preclinical compounds using fixed organoid panels designed for diverse therapeutic applications -
PATIENTS
It's About Time
View the Tempus vision.
- RESOURCES
-
ABOUT US
View Job Postings
We’re looking for people who can change the world.
- INVESTORS
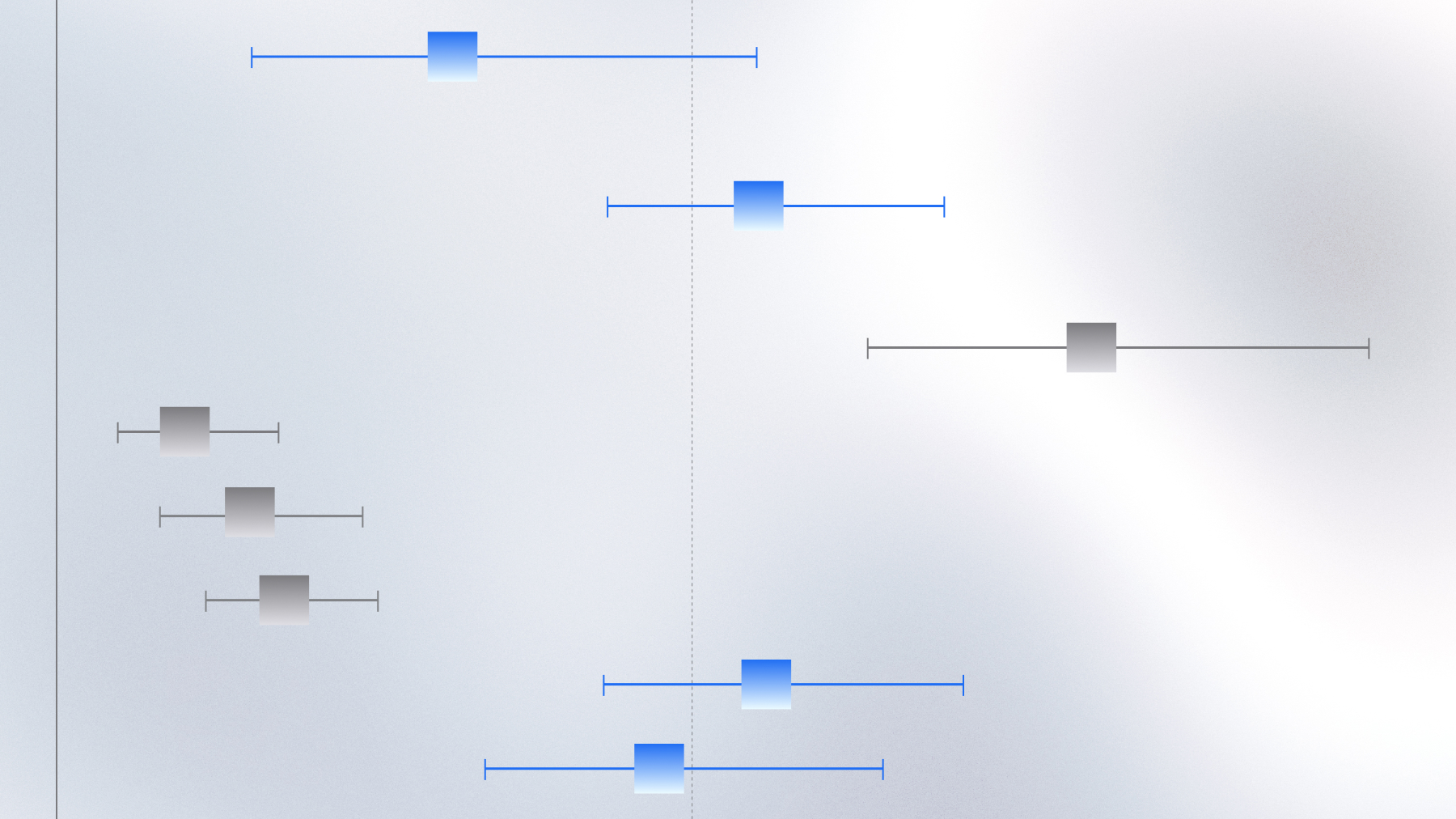
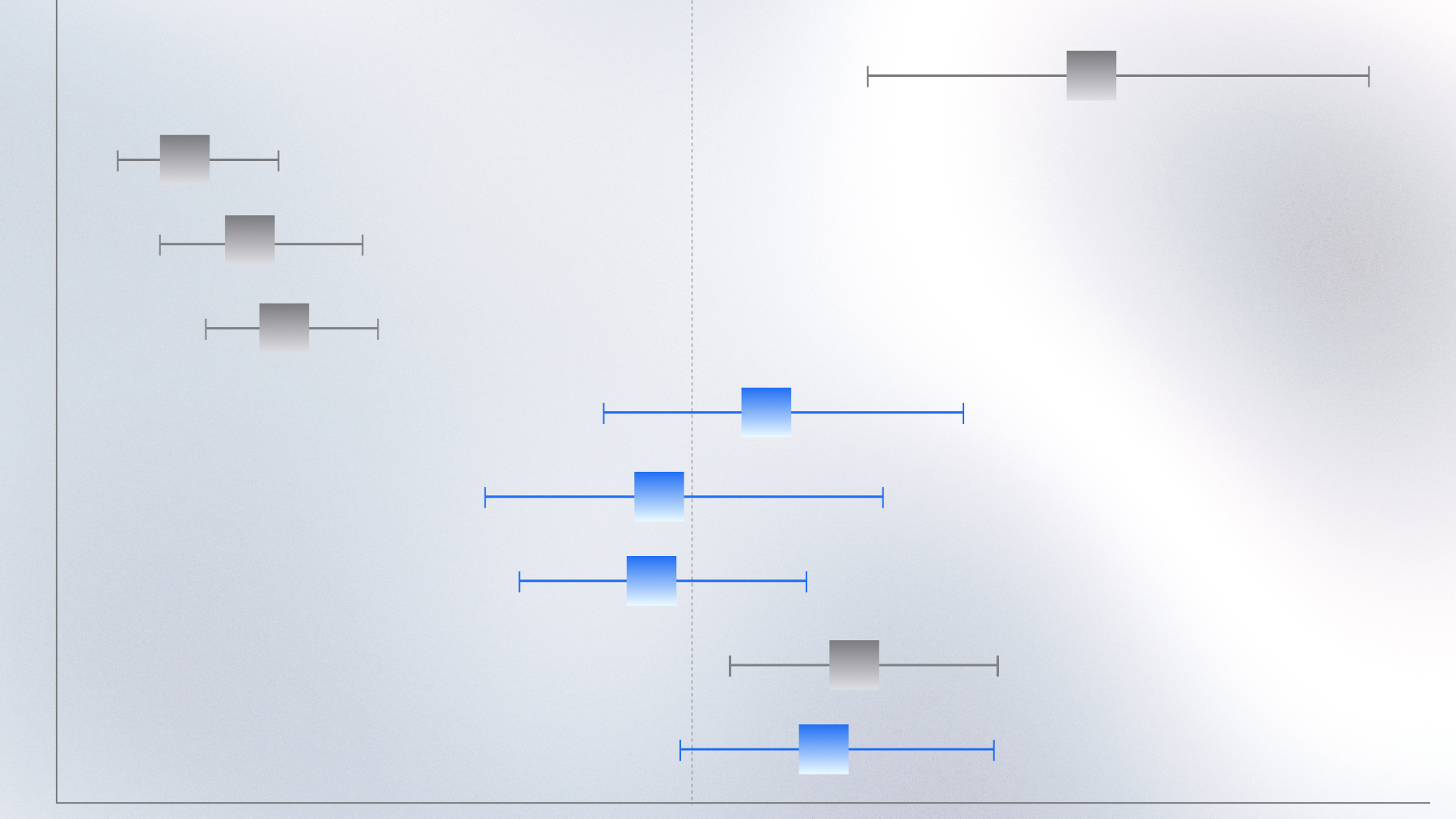
Characterizing KRAS variants in NSCLC with multimodal RWD
RWD analysis reveals distinct immune and metabolic profiles among KRAS-mutant NSCLC
Contact us
Learn more about Tempus or explore more Lens research projects.