-
PROVIDERS
Learn more
Join Tempus at the 2026 ASCO® Annual Meeting!
-
LIFE SCIENCES
Read case study
Learn now Tempus Next Pathways unlocked critical insights into the aNSCLC patient journey with Bristol Myers Squibb (BMS)
-
PATIENTS
It's About Time
View the Tempus vision.
- RESOURCES
-
ABOUT US
View Job Postings
We’re looking for people who can change the world.
- INVESTORS
07/28/2023
Overcoming the Challenges of Variant Calling In PMS2 High Homology Regions For Improved Lynch Syndrome Diagnosis Using Whole-Genome Sequencing
ISMB 2023
PRESENTATION
Authors
Shunhua Han, Vitor Ounchic, Varun Jain, Pavana Anur, Francisco M. De La Vega, James Han
The accurate detection of pathogenic variants in the PMS2 gene is crucial for diagnosing Lynch Syndrome, an autosomal dominant hereditary cancer predisposition syndrome (Lynch et al., 2009). However, the presence of the pseudogene PMS2CL, which includes a ~11kb region of high homology to PMS2 exons 12-15, poses significant challenges for variant calling using short-read next-generation sequencing (NGS, van der Klift et al., 2016). Misalignment and ambiguous mapping of reads originating from this region can lead to false-positive or false-negative variant calls, impacting clinical decision-making and patient care (Huang et al., 2018). In this study, we aim to address these challenges and improve the reliability of small variant calling in the PMS2 high homology region.
Here we explore two alternatives for variant detection in paralogous genes. The first option balances both precision and recall by maximizing the F1 score, while the second emphasizes sensitivity at the cost of precision. In basic or translational research settings maximizing the F-1 score are preferred. In contrast, in clinical diagnostic settings, where identifying all potential pathogenic variants is paramount and confirmatory testing is required (e.g., via long-range PCR-based tests), maximizing recall is preferred, as long as false positive rate is not excessive. Incorporating both methods is important to accommodate the diverse needs of different user groups within the genomics community.
We developed a novel computational method called MRJD (Multi-Region Joint Detection) designed to detect small variants in paralogous regions. Instead of genotyping per individual genomic region, discarding reads with ambiguous alignments, this new method jointly genotypes all paralogous regions using all reads. In addition, we also developed a high sensitivity mode that maximizes the ability to identify all potential variants (Figure 1). These methods are implemented as part of the DRAGEN software suite v4.2, allowing users to choose the one that best fit their need.
We benchmarked MRJD’s variant calling performance on 150 samples from Illumina Polaris diversity panel (>30x coverage using the Illumina NovaSeq 6000 system with PCR-Free library prep and 2x150bp reads, Byrska-Bishop et al, 2022) against orthogonal techniques including long-range PCR NGS approach (Gould et al. 2018). MRJD’s default mode outperforms the DRAGEN default germline small variant caller in the PMS2 high homology region, particularly in terms of INDEL calling. MRJD High Sensitivity mode achieves substantially higher recall compared to other methods, with aggregated recall to be 96% for both SNPs and INDELs (Figure 2). To help interpret the low precision from MRJD High Sensitivity mode, we compared SNPs detected by the MRJD High Sensitivity mode to those identified by the long-range PCR approach (with alleles in PMS2 and PMS2CL all merged into the PMS2 coordinates). The new aggregated precision and recall from MRJD High Sensitivity mode now reaches 91% and 90%, respectively. This analysis suggests that vast majority of false positives from MRJD High Sensitivity mode in the first benchmark analysis are not spurious calls, but rather coming from misplaced variants among paralogous regions or reference difference positions that do not have variants. Finally, we also independently sequenced and benchmarked small variant calling performance on 18 cell line samples from 1000 Genome Project (NovaSeq6000 with 2x150bp PCR free libraries at ~50X depth). Both MRJD modes achieve similar performance as the benchmark analysis using public dataset.
In summary, we introduce a novel computational strategy, MRJD, that addresses the challenge of small variant calling in PMS2 high homology regions using NGS, achieving improved sensitivity and specificity. This work contributes to a more reliable diagnosis of Lynch Syndrome, enabling better risk assessment and personalized management strategies for affected individuals. The MRJD approach is versatile and can be applied to a wide range of segmental duplicated regions. It is estimated that the human genome contains 200-500 medically relevant genes with problematic regions, where high homology is a primary concern (Ebbert et al., 2019; Wanger et al., 2022). We anticipate that our approach will pave the way for further research on variant calling in other medically relevant genes that face similar homology challenges.
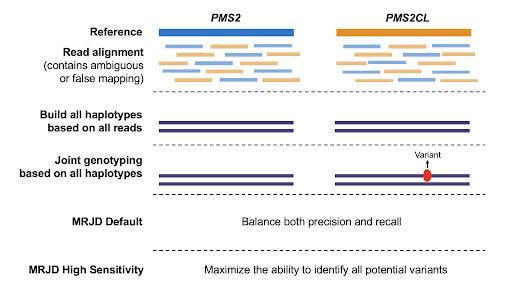
Figure 1. The two workflows of Multi-Region Joint Detection (MRJD) approach.
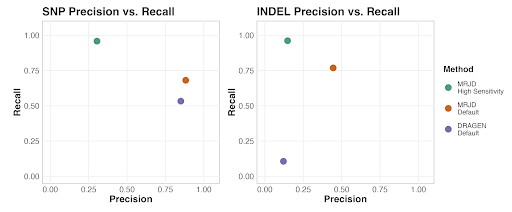
Figure 2. Aggregated SNP and INDEL performance between DRAGEN Default small variant caller, MRJD
Default, and MRJD High Sensitivity mode on 150 samples from Illumina Polaris diversity panel. RTG Tools
ploidy-squash mode is used to generate benchmark statistics.
References
1. Lynch, Henry T., et al. “Review of the Lynch syndrome: history, molecular genetics, screening, differential
diagnosis, and medicolegal ramifications.” Clinical genetics 76.1 (2009): 1-18.
2. van der Klift, Heleen M., et al. “Comprehensive mutation analysis of PMS2 in a large cohort of probands
suspected of Lynch syndrome or constitutional mismatch repair deficiency syndrome.” Human mutation 37.11
(2016): 1162-1179.
3. Huang, Kuan-lin, et al. “Pathogenic germline variants in 10,389 adult cancers.” Cell 173.2 (2018): 355-370. 4.
Gould, Genevieve M., et al. “Detecting clinically actionable variants in the 3′ exons of PMS2 via a reflex workflow
based on equivalent hybrid capture of the gene and its pseudogene.” BMC Medical Genetics 19 (2018): 1-13.
5. Byrska-Bishop, Marta, et al. “High-coverage whole-genome sequencing of the expanded 1000 Genomes Project
cohort including 602 trios.” Cell 185.18 (2022): 3426-3440.
6. Ebbert, Mark TW, et al. “Systematic analysis of dark and camouflaged genes reveals disease-relevant genes
hiding in plain sight.” Genome biology 20 (2019): 1-23.
7. Wagner, Justin, et al. “Curated variation benchmarks for challenging medically relevant autosomal genes.” Nature
biotechnology 40.5 (2022): 672-680.